
Es una condición genética poco frecuente que suelen demorar años en diagnosticarse. Los resultados prometedores de un estudio sobre una opción terapéutica innovadora.
Vivir con una enfermedad poco frecuente (EPOF) muchas veces es una odisea marcada por la incertidumbre, diagnósticos tardíos y la falta de opciones terapéuticas accesibles. En el mundo, una de cada 2.000 personas nace con alguna de estas condiciones, que se caracterizan por su baja frecuencia en la población general. En Argentina, entre el 6% y el 8% de los habitantes—unos 3,5 millones de personas—padecen alguna EPOF.
Este contexto convierte a patologías como la enfermedad de Fabry en un desafío, pero también en un terreno fértil para avances científicos que pueden cambiar la vida de los pacientes.
Infobae accedió a los resultados preliminares del estudio SMILE (Switch Over Study of Biosimilar AGA for Fabry Disease), actualmente en Fase III, que evalúa el primer biosimilar de agalsidasa beta producido en Argentina para el tratamiento de la enfermedad de Fabry. Los resultados revelaron que, tras 26 semanas de tratamiento, se mostró como una alternativa biosimilar segura y efectiva. Los datos completos del estudio estarán disponibles para el primer cuatrimestre de 2025.
Se estima que en Argentina entre 1.200 a 1.500 personas viven con la enfermedad de Fabry. Sin embargo, el médico neurólogo Juan Manuel Politei explicó a Infobae que sólo “unas 900 personas están diagnosticadas y, entre ellas, unas 600 en tratamiento”. El especialista coordinó el estudio SMILE en Argentina y es miembro de la Fundación SPINE, una organización que nuclea a profesionales de la salud dedicados a la asistencia integral de pacientes con enfermedades poco frecuentes.
Cómo se manifiesta la enfermedad de Fabry
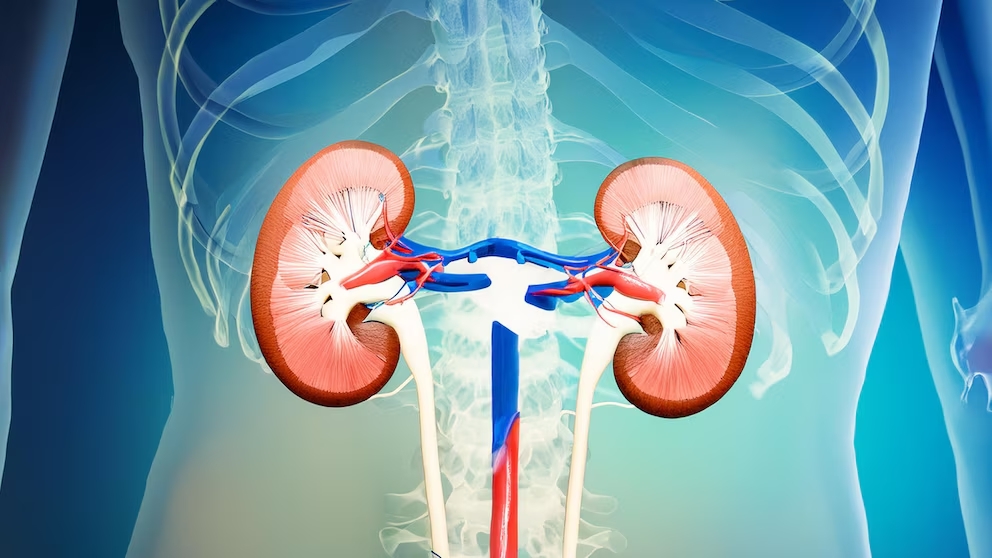
“La enfermedad de Fabry es el resultado de la ausencia genética de una enzima que interviene en la digestión de un grupo de grasas. La ausencia de esta enzima, conocida como alfa-galactosidasa A, lleva a la acumulación de glicolípidos dentro de un compartimento de las células que se llaman lisosomas. Esta acumulación genera daño en distintos órganos, que puede progresar a insuficiencia renal y cardíaca, entre otras complicaciones”, comentó el doctor Politei.
Esto hace que la enfermedad de Fabry sea una condición multisistémica que requiere un manejo temprano y multidisciplinario. Si bien en los últimos años es menor el tiempo que demora identificar la condición, sigue siendo una enfermedad de difícil diagnóstico y los pacientes puede deambular entre consultas a distintos especialistas sin lograr ponerle “nombre y apellido” a los síntomas que padecen.
El diagnóstico de la enfermedad de Fabry puede demorar unos 8 años en promedio, debido al desconocimiento general sobre la enfermedad y a la inespecificidad de sus síntomas iniciales, como dolores en las extremidades, problemas gastrointestinales recurrentes y lesiones cutáneas.

Este retraso puede derivar en complicaciones graves e irreversibles, como insuficiencia renal, fibrosis cardíaca o accidentes cerebrovasculares, afectando drásticamente la calidad de vida de quienes la padecen.
Aunque se considera una enfermedad poco frecuente, estudios recientes sugieren que la incidencia podría ser mayor a la estimada. Se calcula que uno de cada 40,000 varones recién nacidos podría padecerla, aunque las mujeres también pueden desarrollar síntomas, generalmente menos severos, debido al patrón hereditario ligado al cromosoma X.
Cómo es el tratamiento de la enfermedad de Fabry
El tratamiento estándar para la enfermedad de Fabry se basa en la terapia de reemplazo enzimático (TRE), que consiste en la administración intravenosa de la enzima alfa-galactosidasa A, cada 14 días. Este procedimiento busca compensar la ausencia de la enzima en los pacientes, al ayudar a reducir la acumulación de globotriaosilceramida (GL-3) en los tejidos, lo que contribuye a mitigar los síntomas y retrasar la progresión de complicaciones graves, como insuficiencia renal y cardíaca.

Además, en ciertos casos, particularmente en las formas tardías de la enfermedad, es posible utilizar una terapia oral que mejora la funcionalidad de la enzima cuando el cuerpo logra producir pequeñas cantidades. Esta opción beneficia aproximadamente al 30% de los pacientes.
En este contexto, el laboratorio argentino Biosidus logró un avance con el desarrollo de un biosimilar de agalsidasa beta, una de las enzimas utilizada en la terapia de reemplazo enzimático (TRE). Este tratamiento es el primer biosimilar de este tipo desarrollado y producido en Argentina
Según los resultados preliminares del Estudio SMILE el biosimilar demostró seguridad y eficacia tras 26 semanas de tratamiento. “Es un logro nacional muy importante”, destacó el doctor Politei, quien fue coordinador del estudio a nivel local.
“Como investigador, noté, el beneficio personalmente, no lo leí en un artículo. El estudio se realizó en el país, con pacientes argentinos, en centros de investigación de las provincias de Buenos Aires, Córdoba y La Rioja. Este biosimilar cumple con todas las normativas y criterios regulatorios, fases clínicas y preclínicas, y ha demostrado seguridad y eficacia”, enfatizó el neurólogo.

El biosimilar de agalsidasa beta fue investigado a través de un programa de ensayos preclínicos. En estos estudios, se observó una alta similitud con el tratamiento de referencia en términos de propiedades fisicoquímicas y biológicas. Además, se comprobó que el perfil farmacocinético, farmacodinámico, inmunogénico y de seguridad del biosimilar es comparable al del medicamento innovador. Los datos de seguridad obtenidos hasta el momento en el Estudio SMILE también respaldan esas conclusiones.
Al tratarse de un biosimilar, se espera que su costo sea considerablemente menor, lo que permitiría aumentar la cobertura a más personas, ampliando el acceso a terapias de reemplazo enzimático.
“Es la primera vez que se realiza un estudio de estas características en el país y llevar adelante este estudio de investigación clínica localmente, que no tiene nada que envidiarle a uno equivalente hecho por cualquier otro laboratorio de envergadura multinacional, nos aporta un diferencial robusto. Alcanzar este resultado es un hito para la biotecnología de las enfermedades raras”, afirmó la doctora Viridiana Berstein (MN 118.280), médica especialista en investigación clínica y gerente de investigación clínica del Laboratorio Biosidus.
Cuáles son los síntomas de la enfermedad de Fabry

La enfermedad de Fabry es un claro ejemplo de los desafíos que enfrentan las Enfermedades Poco Frecuentes (EPOF), causada por una deficiencia de la enzima alfa-galactosidasa (alfa-GAL), resulta en la acumulación de un lípido llamado globotriaosilceramida (GL3) en múltiples órganos, causando daños progresivos.
Existen dos tipos de enfermedad de Fabry, la forma clásica y la forma tardía. En la primera, que aparece entre los 4 y 6 años, hay ausencia total de la enzima alfa-galactosidasa A. Esto provoca crisis de dolor en manos y pies, desencadenadas por fiebre, ejercicio o cambios de temperatura, además de dolor abdominal, diarrea y distensión. En la adolescencia, se suman problemas de sudoración y lesiones cutáneas llamadas angioqueratomas. La forma tardía, en cambio, se manifiesta entre los 30 y 40 años debido a una producción parcial de la enzima, con síntomas focalizados en el corazón o los riñones.
Se trata de una condición multisistémica, que afecta principalmente al sistema nervioso periférico, los riñones, el corazón, el sistema gastrointestinal y el cerebro. Los síntomas iniciales incluyen sensación de quemazón y hormigueo en manos y pies (acroparestesias), alteraciones gastrointestinales (dolor abdominal, diarrea o estreñimiento), y lesiones en la piel (angioqueratomas) o en la córnea (córnea verticillata). Estos síntomas suelen aparecer durante la niñez, pero con frecuencia son malinterpretados como trastornos psicológicos o relacionados con el crecimiento.
En adultos, la acumulación progresiva de GL3 puede causar hipertrofia miocárdica, insuficiencia renal e incluso ACV precoces, complicaciones que suelen manifestarse en la tercera o cuarta década de vida.
Además, al tratarse de una enfermedad hereditaria, el subdiagnóstico impacta a familias enteras. Es habitual que el diagnóstico de un paciente permita identificar casos en parientes, por eso los especialistas insisten en la importancia de implementar estudios genéticos y elaborar árboles genealógicos para la detección precoz.
En relación con la incidencia, se estima, con base en datos internacionales, que la forma clásica de la enfermedad de Fabry afecta, en promedio, a 1 de cada 20.000 recién nacidos vivos, mientras que la forma tardía se presenta en 1 de cada 3.000, aunque estas cifras pueden variar entre países.
En la Argentina, se considera enfermedad poco frecuente (EPOF) a aquellas patologías cuya prevalencia en la población es igual o inferior a 1 persona cada 2.000 habitantes; esto es lo que establece la Ley N° 26.689 de “Cuidado integral de la salud de las personas con EPOF y sus familias”, promulgada en junio de 2011 y reglamentada en 2015.
Frente a este panorama, la aparición de un nuevo tratamiento siempre es una buena noticia para los pacientes y sus familias. Se estima que una vez completados los análisis secundarios del Estudio SMILE, el biosimilar para el tratamiento de la Enfermedad de Fabry esté disponible en Argentina en el segundo semestre de 2025, con perspectivas de exportación a otros países emergentes y desarrollados.
Fuente: Infobae



